Notre équipe vise à décrypter comment ApoE4 contribue à la dégénérescence synaptique associée à la pathologie tau dans la maladie d'Alzheimer en : 1) identifiant, au niveau des synapses endommagées par tau, les modifications synaptiques fonctionnelles et structurelles induites par ApoE4 ; 2) identifiant les protéines responsables de ces changements structurels et fonctionnels ainsi que leur origine cellulaire ; 3) concevant des traitements ciblant ApoE ou ses effecteurs en aval pour favoriser le bien-être synaptique dans la maladie d'Alzheimer. Cibler les dysfonctionnements synaptiques devient une stratégie thérapeutique attrayante car ils se produisent aux premiers stades de la maladie et la préservation des synapses est cruciale pour la fonction cérébrale et la cognition. Cependant, la lutte contre la perte synaptique a été relativement négligée jusqu'à présent en raison de la méconnaissance des mécanismes synaptotoxiques (défi scientifique) et de la difficulté à atteindre une résolution synaptique (défi technologique). Notre équipe vise à relever ce défi scientifique en étudiant le rôle du facteur de risque génétique le plus puissant dans la maladie d'Alzheimer (ApoE4) dans la dégénérescence synaptique associée à la pathologie tau, en tenant compte du rôle des cellules gliales. Nous visons également à surmonter l'obstacle technologique en atteignant une résolution synaptique expérimentale de haut niveau en utilisant la protéomique avancée, de la chimigénétique et de la microscopie super-résolution au niveau synaptique, associée à l'électrophysiologie et aux tests comportementaux.

La maladie d'Alzheimer (MA) est l'un des plus grands défis socio-économiques du XXIe siècle pour lesquels il n'existe actuellement aucun remède. La MA se caractérise par l'accumulation de la protéine amyloïde-β (Aβ) en plaques et l'agrégation de la protéine tau dans les neurones (pathologie tau). Dans la MA, l'accumulation progressive de la pathologie tau conduit à la dégénérescence des synapses (zone de contact entre deux neurones où a lieu la transmission du message nerveux) et, par conséquent, aux troubles de la mémoire bien connus dans la MA.
L'apolipoprotéine E (ApoE) est une protéine essentielle pour les fonctions synaptiques. L'isoforme E4 d'ApoE (ApoE4) augmente fortement le risque de développer Alzheimer. Bien que nos études précédentes montrent qu'ApoE4 aggrave la perte synaptique, nous ne savons pas comment.
Notre travail vise à caractériser avec une résolution synaptique comment ApoE4 contrôle la dégénérescence synaptique associée à la pathologie tau dans la MA, et à utiliser ces nouvelles connaissances pour concevoir des stratégies thérapeutiques efficaces contre cette maladie incurable.
Pour cela, nous avons développé trois axes de recherche :
1. Changements synaptiques : Un premier axe de recherche de l'équipe vise à identifier comment ApoE4 modifie les fonctions et les structures des synapses endommagées par tau.
2. Acteurs cellulaires et protéiques : Un deuxième axe de recherche vise à déterminer si ApoE4 requiert la microglie (cellules immunitaires du cerveau) et/ou les astrocytes (fournissant un soutien métabolique et physique aux neurones) pour aggraver la perte synaptique dans la MA. De plus, nous visons à identifier les protéines responsables de la perte synaptique au niveau des synapses, ainsi que l'origine cellulaire de ces protéines (neurones, astrocytes ou microglie).
3. Traitements : Enfin, notre dernier axe de recherche vise à identifier de nouvelles cibles thérapeutiques ciblant ApoE4, mais aussi les acteurs protéiques et cellulaires identifiés dans le deuxième axe de recherche, afin de protéger les synapses dans la MA.
Ces trois sujets de recherche aideront à mieux comprendre les relations entre la microglie, les astrocytes et la pathologie tau conduisant à la perte synaptique, dans le but de trouver de nouvelles cibles thérapeutiques pour protéger les synapses dans la maladie d'Alzheimer.
Déterminer avec une résolution synaptique sans précédent comment ApoE4 aggrave la dégénérescence synaptique associée à la pathologie tau dans la MA nécessite une caractérisation fine des fonctions d'ApoE4 au niveau des synapses endommagées par tau. Cette stratégie est cruciale pour valider la synapse en tant que cible pour le développement de stratégies innovantes visant à favoriser le bien-être synaptique dans la MA.
Notre équipe émet l'hypothèse qu'ApoE4 contribue à la dégénérescence synaptique associée à la pathologie tau en altérant les interactions entre les cellules gliales et les neurones.
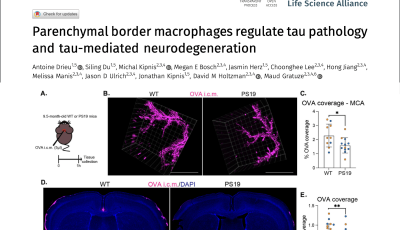
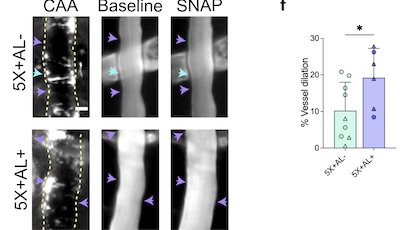
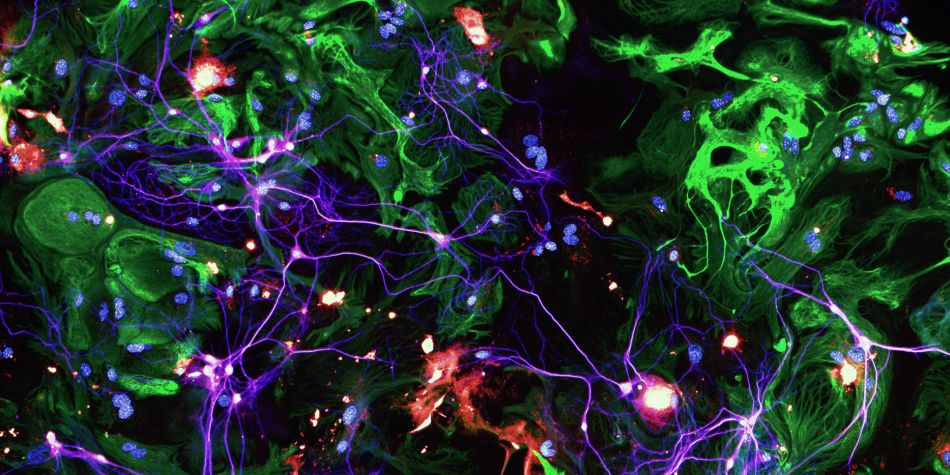
1 : CHANGEMENTS SYNAPTIQUES : Des études in vivo ont clairement montré qu'ApoE4 aggrave les pathologies associée à la pathologie tau (Gratuze et al., 2023 ; Shi et al., 2017 ; Chao Wang et al., 2021 ; Zhao et al., 2020). Dans une étude récente, j'ai rapporté qu'ApoE4 diminue la protéine PSD95 post-synaptique chez les souris P301S (Gratuze et al., 2023), suggérant qu'ApoE4 aggrave la dégénérescence synaptique associée à la pathologie tau. En raison du rôle d'ApoE dans les fonctions synaptiques et dans la MA, nous visons à déterminer comment les isoformes d'ApoE affectent la dysfonction synaptique associée à la pathologie tau. Nous cherchons à identifier les changements synaptiques fonctionnels et structurels causés par ApoE4 au niveau des synapses endommagées par tau, en présence ou non de microglie, en combinant la microscopie super résolution, l'électrophysiologie et des tests comportementaux.
2 : ACTEURS PROTÉIQUES : Nous avons précédemment montré qu'ApoE4 aggrave les altérations synaptiques chez les souris P301S, mais il reste à déterminer quelles protéines sous-tendent ce phénomène, leur origine cellulaire et leur dépendance vis-à-vis des interactions moléculaires entre les types cellulaires (c'est-à-dire les neurones et les astrocytes). Comprendre les mécanismes pathogènes derrière l'axe ApoE4-tau est crucial pour élucider le rôle d'ApoE dans les altérations synaptiques, et par extension également le rôle des astrocytes, car ils sont la principale source d'ApoE. En conséquence, nous évaluons comment les isoformes d'ApoE affectent le protéome naissant dérivé des astrocytes ou des neurones (c'est-à-dire les protéines nouvellement synthétisées) au niveau des synapses. De plus, nous évaluons les changements spécifiques aux isoformes d'ApoE à l'interface astrocyte/synapses en utilisant une approche chimigénétique in vivo basée sur la complémentation de fragments de surface cellulaire. Nous cherchons à identifier les acteurs protéiques sous-jacents aux changements structurels et fonctionnels associés à ApoE4 décrits dans le premier sujet de recherche, en combinant des approches chimigénétiques et des protéomiques avancées pour taguer et quantifier spécifiquement les protéines produites par les neurones ou les astrocytes au niveau : 1) des synapses, et 2) de l'interface entre les astrocytes et les synapses.
3 : TRAITEMENT : La maladie d'Alzheimer est un grave problème de santé publique avec environ 50 millions de personnes touchées dans le monde, soulignant l'urgence de développer des stratégies pour ralentir/prévenir/guérir la MA. À cet égard, on estime qu'un traitement retardant l'apparition de la maladie de 5 ans réduirait le nombre de cas de moitié. En raison du rôle d'ApoE4 dans les altérations synaptiques et la neurodégénérescence associée à la pathologie tau, nous proposons de développer et de tester des thérapies ciblant 1) ApoE et les récepteurs d'ApoE, et 2) les acteurs protéiques les plus prometteurs au niveau des synapses endommagées par tau identifiés dans le deuxième sujet de recherche pour protéger les synapses dans la MA.
![]()

![]()

![]()

![]()